Dr. Geoffrey Lowman
Senior Staff Scientist, Molecular Biology, Clinical Sequencing Division
Thermo Fisher Scientific
What is your group’s role in research and development for the clinical sequencing division?
For the past 5 years our research group has focused on developing panels to sequence B or T cells. Our focus is the immune repertoire with its many applications from immuno-oncology biomarker discovery to precision oncology research. It’s a very rich field of study.
What is immune repertoire sequencing and why is it important?
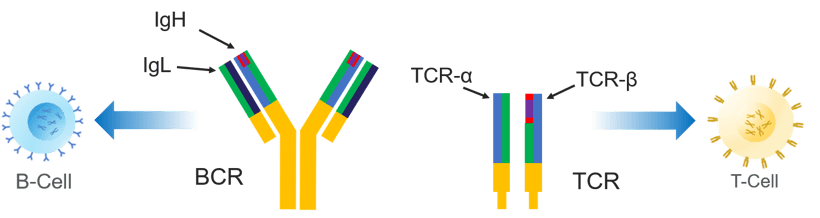
Immune repertoire sequencing in this context encompasses the diverse array of antigen receptors expressed in the population of B and T cells contained in a sample. These cells are part of the adaptive immune system which the body uses to fight off foreign antigens, and anything else that might be attacking the body. We use targeted next-generation sequencing (NGS) to get a detailed portrait of the repertoire of B or T cells in a particular sample.
During B- and T-cell development, a portion of the genome is rearranged to be able to express receptors on the cell surface, including the B-cell receptor (BCR) and T-cell receptor (TCR), respectively. Each rearrangement is unique for that B or T cell. It's like a molecular barcode that can be used to identify an individual cell clone.
The panels that we develop sequence the portion of the genome that's rearranged to form those B- and T- cell receptors, including the different chains and subunits that comprise each receptor. Through sequencing we can identify B- and T-cell clones and take a closer look at how the frequencies of those clones relate to what's going on in a sample. We can also drill down further to compare the individual nucleotide sequences of clones.
What are the applications of BCR and TCR sequencing?
The data gained is particularly relevant for lymphoid malignancy research, including the study of leukemias, lymphomas, and multiple myeloma. One application where sequencing has really taken hold is clonality detection, which refers to the expansion of a B or T cell. In the context of oncology research, we're looking for a particular B or T cell that has expanded.
In lymphoma or leukemia samples, you'll often find a dominant clone that has significantly expanded relative to the other cells in the repertoire. We're sequencing B- or T- cell receptors with NGS panels to gain more insight for research into the disease process. Being able to identify the clone and read the full sequence informs oncology research.
What are the benefits of NGS over traditional methods?
The benefits of NGS are clear when it comes to sensitivity or limit of detection. While older methods such as flow cytometry or capillary electrophoresis may be great at identifying clonal samples, you don't get the full clone sequence information. You gain a good deal of information with NGS and its low limit of detection.
NGS can typically detect 1 cell in a million (10-6), which is 100-fold more sensitive than traditional methods like flow cytometry. It means that you can look at a study endpoint and see what’s happening at those low limits of detection. To be able to read the sequence at 1 cell in 1 million as an endpoint in a research trial using NGS is unprecedented when compared to traditional methods.
Are there additional benefits to having the entire sequence of a clone?
Deeper insights into clonal evolution inform research. You can determine two related clones in the same sample are members of the same clonal lineage. Lineage analysis is automated in our analysis software in Ion Reporter.
For instance, if you're conducting an experiment studying late-stage B cell samples, our lineage analysis tool can identify a clone of interest and other clones of the same lineage. Perhaps this clone contains ongoing somatic hypermutation. This is critical information as we expand the way we can interpret the process by which these cells expand in frequency. Our new assays incorporate primers for multiple receptors in a single library reaction to provide more sequences to analyze for clonality detection, potentially increasing the probability of identifying a clone of interest and delves deeper to identify clonal lineages.
Can you discuss the primer design strategy for the new Pan-Clonality assays?
Our immune repertoire assays utilize AmpliSeqTM library preparation technology, which has been part of the Ion platform for over a decade.
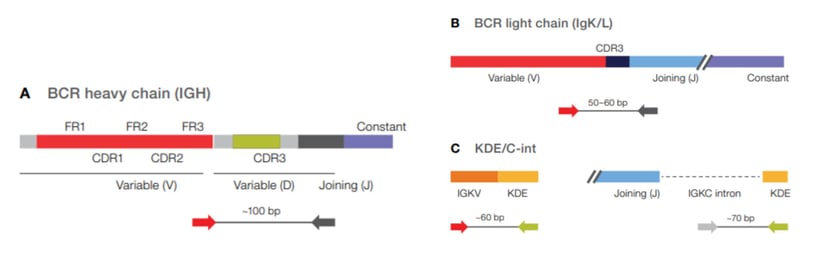
Our library prep technology enables us to reduce primer dimer in highly multiplexed reactions. Consequently, you can use multiple primers in a single library prep reaction with less primer–primer interaction or primer bias. That's the key to enabling assays for multiple immune-cell receptors in a single reaction. For example, in our OncomineTM BCR Pan-Clonality Assay, we include primers for the heavy chain (IGH), for both light chains, kappa (IgK) and lambda (IgL), as well as the kappa deletion element (KDE) and c-intron (Cint)—all in a single tube reaction.
The amplicons generated by the BCR Pan-Clonality Assay are in a similar length range and they will perform well throughout the templating and sequencing workflow on the Genestudio™ and Ion Chef™ platforms. To my knowledge, no other commercial assays can determine clonality in multiple receptors with a single library reaction.
What's the functional benefit of having multiple receptor targets in a single reaction?
If you're determining if a sample is clonal or not, your goal is to find primers to amplify the expanded clone. For example, if you only use a heavy chain primer set and the heavy chain has undergone extensive somatic hypermutation, there may be cases where mutations occur under primer binding sites.
Consequently, you may not detect a clone in your initial attempt. Thus, our BCR Pan-Clonality Assay includes multiple primer sets—to simultaneously target different areas of the receptor in a single reaction, increasing the probability of clone detection. The BCR Pan-Clonality assay, for example, includes the heavy chain, kappa chain, lambda chain, and the kappa deletion element primers—that's 4 assays in 1 tube. In both external and internal studies, we saw positive clonality detection rates in the range of 93% to 95% using a single library reaction. This is a significant improvement from what you would expect from a single heavy chain assay alone.
Why did you choose to include multiple light chain targets in the BCR Pan-Clonality assay?
In B-cell biology, the kappa chain would be a primary target because the lambda chain is only expressed after the kappa chain is deleted. A small sequence remains from that kappa deletion event. Some assay developers believe that you should be able to detect clones with only primers for the kappa region and the kappa deletion element. This is based on the premise that everything with a lambda chain should have the kappa deletion element present. However, we might assume in these afflicted cells that standard B-cell biology is altered.
We have a couple of examples from our development work in which we detected clonality only with the lambda light chain primers but not the kappa deletion element. Without including the lambda chain, a researcher might not be able to detect the clone, leading to a false negative result.
Can you address the need for secondary testing in the event of a test failure?
When you can't detect a clone, secondary testing allows you to eliminate the possibility that you're missing something due to primer failure, for instance due to somatic hypermutation at a primer binding site. You would suspect some degree of primer failure along a gene where somatic hypermutation is so prevalent, particularly in late-stage B cell samples. Including more receptors helps us minimize the need for secondary testing, as we regularly attain a 93-95% detection rate with our single-tube assay.
However, we do offer secondary assays. One of these secondary assays primes in the distal region of framework 3 (FR3) of the IGH variable gene, which is unique. We've seen solid performance with 70% to 80% IGH clonality detection across various cell lines and clinical research samples using our Oncomine IGH FR3(d)-J assay.
How does the sensitivity of these assays compare with traditional approaches?
With traditional methods such as flow cytometry, the limit of detection is typically in the range of 10-4. qPCR methods can go lower, but the main drawback is that you must design specific probes for whatever sequence you're priming. The lowest range you can hope to achieve with the best of traditional methods is 10-5.
With NGS, reaching that limit of detection (1 in 100,000 or 10-5) is standard. Our new assays don't sacrifice sensitivity just because we have primers for multiple receptors. Our standard limit of detection is 10-5 and can reach as low as 10-6. A limit of detection of 10-5 is a benchmark that makes NGS economically viable for these applications, and 10-6 is what's needed to thoroughly validate an assay for application at a measurement level of 10-5. Our new assays (both primary and secondary) are internally validated to detect known arrangements at 10-6.
What is B-cell receptor somatic hypermutation?
Mutations in B cell receptors are far more numerous than any other portion of DNA in the genome. These high levels of mutation are due in part to the receptor improving its affinity for antigen. It starts when an antigen stimulates a naive B cell. The B cell then goes through the germinal center in secondary lymphoid tissues, engaging in the process of affinity maturation where the receptor mutates in order to increase its affinity for a particular antigen. Finally, a B cell with the improved affinity will survive to another round of affinity maturation and so on.
The mutation rate, or the number of mutations, is something that we can measure by sequencing the B-cell receptor and then calculating the mutation rate by comparing the results against a known germline reference sequence.
Ongoing somatic hypermutation can be used to analyze a B-cell lineage. For example, given 2 clones, 1 of which undergoes somatic hypermutation – you now have 2 related clones (a clonal lineage) with slightly different sequences. Lineage analysis is completely automated in our analysis pipeline and is reported in a lineage table.
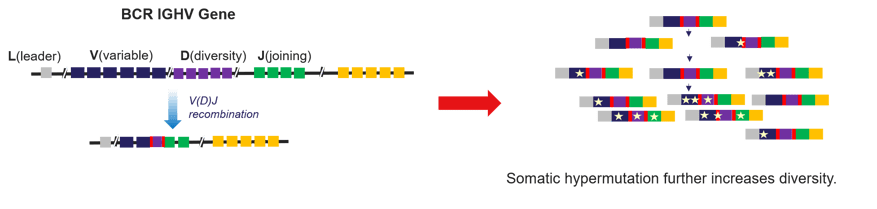
Somatic hypermutation involves the introduction of point mutations along the BCR IGHV gene, which increases repertoire diversity and increases antigen affinity.
What are the implications of somatic hypermutation in clinical research applications?
When measuring somatic hypermutation, you want to be as accurate as possible, which means you want to cover the entire IGH heavy chain sequence if possible. The European Research Initiative on CLL or ERIC, recommends that assays prime the IGHV leader region — providing sequence across the entire functional receptor. This distance spans a sequence of about 500 base pairs.
It's very long, but it gives you an accurate readout of the mutation rate across the chain. That's important because it allows you to determine what stage of maturation a B cell is at in a particular CLL sample. There are 2 broad categories: unmutated (< 2% mutation rate), and mutated (> 2% mutation rate). We have designed a Leader-J assay to accurately measure somatic hypermutation in accordance with ERIC recommendations.
Is substitution error rate a concern when measuring somatic hypermutation?
When trying to measure a series of point mutations, the accuracy of the sequencing technology is critical. A high error rate can make it very challenging to differentiate a single-point mutation from a single-base substitution error.
There is a great deal of randomness and diversity in the DNA following genomic rearrangement to form B- and T-cell receptors. If you have a high substitution error rate, you will likely see some low-frequency clones that appear to be related to a high-frequency clone which aren't due to somatic mutation, but instead stem from substitution errors in sequencing.
Fortunately, the Ion TorrentTM platforms are based on semiconductor sequencing, and using natural nucleotides in our chemistry leads to a low base-substitution rate—an advantage for immune repertoire sequencing in particular. In addition, with our long-read amplicon, we can confidently measure somatic hypermutation with less concern for the substitution error rate that may be present on other platforms.
Were there any special design considerations for the workflow and analysis for these assays?
Yes, we thought about the entire user experience from a fully integrated workflow and practical analysis perspectives. We wanted to help simplify the complexity of immune repertoire sequencing for our customers. We didn’t want them to worry about building the back-end analysis tools for these assays.
The manual library prep workflow is familiar to most of our customers as the Ion sequencing platform is well established.
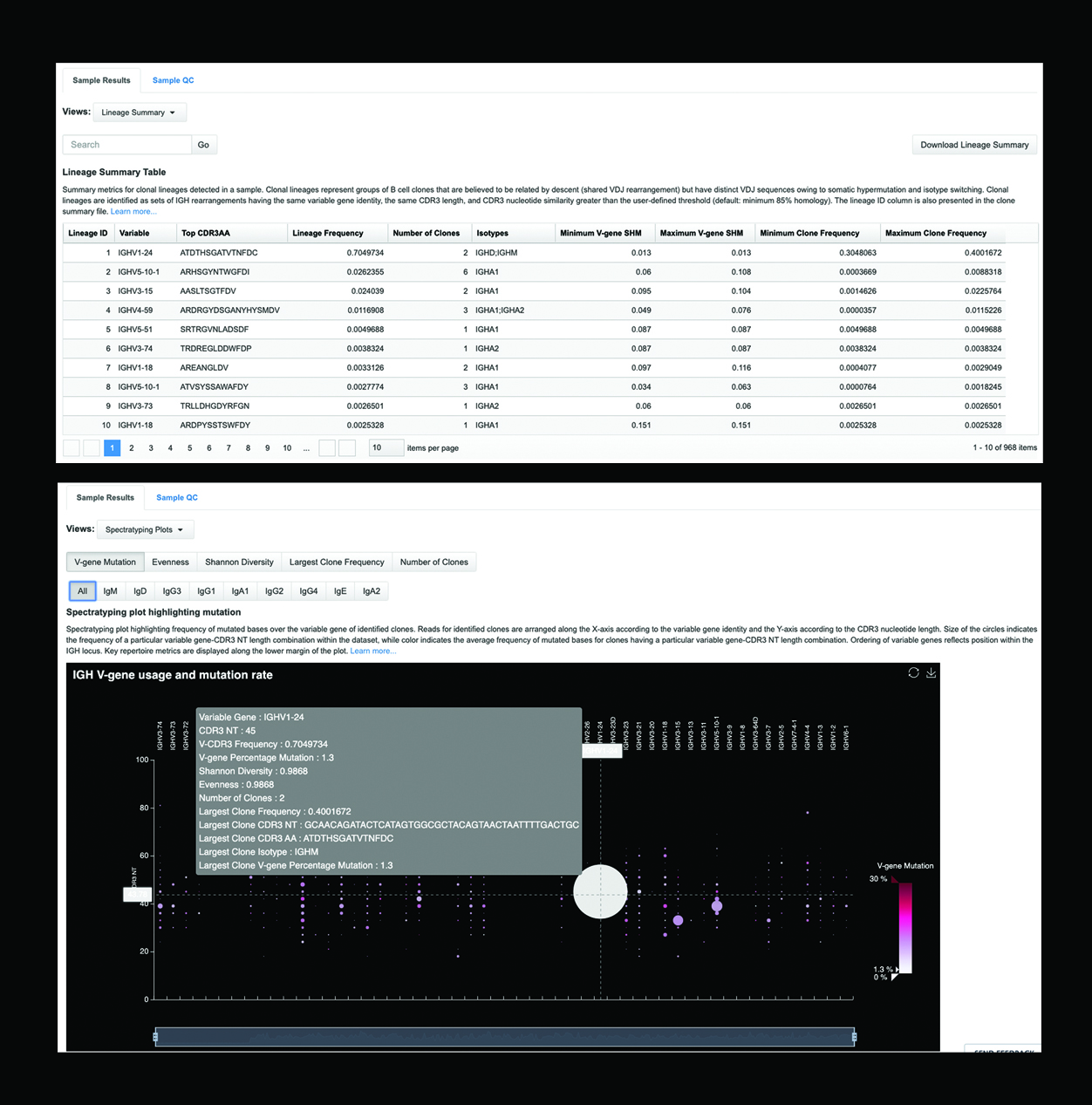
Our goal was to make the analysis user-friendly. We built practical features and powerful visualization tools so even those who aren’t immunologists can glean the information they need. Democratizing sequencing has been our mission since the advent of the Ion platform. Our spectratyping plot tools graphically represent clonal expansion and other features in the repertoire at a glance. Other visualizations in our analysis output cover variable gene diversity and joining gene diversity within the repertoire, CDR length histograms, and more. There is a clone table that clearly reports every clone’s variable gene identity and joining gene identity, amino acid sequence, nucleotide sequence, and clone frequency.
As mentioned in our earlier discussion, you can access a lineage table with any of our B-cell analysis tools. The results from all tables are also downloadable, as we know people like to create their own visualizations and perform their own secondary analyses. A downloadable metrics file lets you dig into many other repertoire metrics. It’s a robust and highly developed analysis solution created entirely with our customers’ needs in mind.
Learn More: See our Immune Repertoire Assays For Hemato-Oncology Research.


Precision medicine is rapidly changing our understanding of cancer research and treatment decisions. These breakthrough, personalized treatments hold promise even for patients with historically hard-to-treat diseases, like lung or breast cancer. But
In his recent talk at OncomineWorld 2023, Dr. Bekim Sadikovic presented a strong, evidence-based argument for frontline next-generation sequencing (NGS) in myeloid malignancy testing.
Bekim Sadikovic PhD, DABMGG, FACMGProfessor, Research Chair, and...
In a recent Association for Molecular Pathology (AMP) workshop, Dr. Cecilia Yeung shared how her lab is implementing rapid next-generation sequencing (NGS) to help advance hemato-oncology research and expand access to underserved populations.
...